Поговорим о том, что такое талассемия
Гемоглобин – элемент эритроцитов, его задача – транспортировать кислород. В самом общем виде кровь состоит из альфа- и бета-белков. Эритроциты не производятся в нужном количестве и не могут перенести достаточный объем кислорода, когда организм не вырабатывает удовлетворительное число одного из этих белков. Результатом является анемия, развивающаяся с рождения и длящаяся на протяжении всей жизни.
Талассемия – название не одного расстройства, это целая группа заболеваний, которые по-разному влияют на организм человека.Содержание
Классификация
Различают такие виды, как α- и β-талассемию.
Альфа-талассемия
 В формировании альфа-цепи гемоглобина участвуют 4 гена (по 2 от каждого из родителей).
В формировании альфа-цепи гемоглобина участвуют 4 гена (по 2 от каждого из родителей).
- Если мутация в 1 гене — симптомов болезни нет. Но человек является носителем заболевания, есть вероятность того, что он передаст болезнь ребенку;
- Мутировало 2 гена – лёгкая форма заболевания (малая альфа-талассемия, гетерозиготная талассемия);
- 3 гена – проявление симптомов будет умеренным. Эта форма называется гемоглобинопатия H (hemoglobin H disease);
- Четыре гена – альфа-талассемия майор (alpha-thalassemia major, hydrops fetalis). Состояние приводит к выкидышу, или ребенок погибает почти сразу после родов.
Бета-талассемия
 При талассемии этого вида в формировании бета-цепи участвуют 2 гена (один от каждого родителя).
При талассемии этого вида в формировании бета-цепи участвуют 2 гена (один от каждого родителя).
- Мутация одного гена – малая бета-талассемия (beta-thalassemia minor,beta-thalassemia trait). Лёгкая форма заболевания.
- Мутация двух генов свидетельствует о том, что проявления болезни будут сильными. Это бета-талассемия майор (большая талассемия, beta-thalassemia major) или анемия Кули (Cooley’s anemia). Дети такими генами здоровы в первые дни жизни, но в последующие два года у них развиваются симптомы болезни.
Причины заболевания
Талассемия проявляется из-за мутаций в ДНК клеток, которые составляют гемоглобин. Эти мутации нарушают нормальную производительность гемоглобина и влекут за собой снижение его уровня, вызывая анемию. Это генетический дефект, наследуемый ребенком от родителей. Если один из родителей – носитель болезни, у ребенка, скорее всего, разовьется талассемия минор. Видимых симптомов не будет, но ребенок станет носителем заболевания. Некоторые люди с малой талассемией замечают незначительные симптомы.
Про талассемию и риски заболеть в видео ниже
Если оба родителя являются носителями талассемии, есть больший вероятность унаследовать тяжелую форму заболевания.Симптомы и признаки
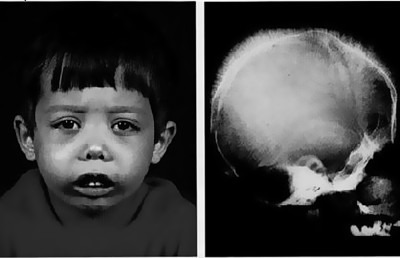 Повышенная усталость;
Повышенная усталость;- Слабость во всём теле (из-за снижения уровня гемоглобина);
- Бледность кожных покровов, изменение цвета кожи на жёлтый (на фото имеет ярко-жёлтый оттенок);
- Дефекты лицевого скелета;
- Замедленное развитие;
- Скопление газов, вздутие, метеоризм;
- Темно-коричневая моча.
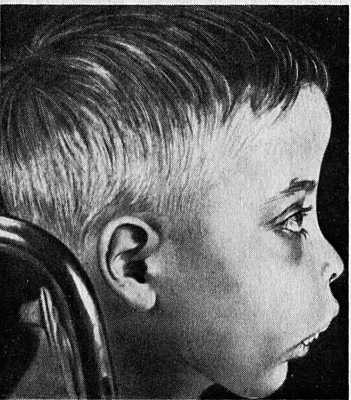 Признаки и симптомы разных типов болезни можно дополнить следующими:
Признаки и симптомы разных типов болезни можно дополнить следующими:
- Альфа-талассемия майор: бледность, вздутый живот, явные гематологические нарушения у новорожденных;
- Бета талассемия минор: состояние гемолитической анемии, сильная бледность, вздутый живот из-за гепатоспленомегалии;
- Резкие костные изменения из-за неэффективного производства эритроцитов (гиперостоз лобной кости, видимые лицевые кости, изменение зубного прикуса);
- Гиперметаболизм;
- Подагра в связи с гиперурикемией (редко);
- Слишком большое количество железа;
- Замедление роста и развития;
- Метаболические симптомы (сахарный диабет, заболевания щитовидной железы);
- Невропатия / паралич у больных с тяжелой анемией.
Диагностика
Проводится путём проведения исследований, таких как диагностика костного мозга, электрофорез гемоглобина, анализ крови на гемоглобин. Общего анализа крови, как правило, достаточно, чтобы начать подозревать болезнь.
Задача генетических исследований семьи – показать, является ли пациент носителем.Пренатальная диагностика проводится на 11 неделе беременности путем взятия пробы части плаценты. Плод может быть протестирован при помощи анализа околоплодных вод (амниоцентеза) на 16-й недели беременности. Во время процедуры используется специальная игла, чтобы взять пробу жидкости, окружающей ребенка, для тестирования.
Существует новая методика предимплантационной генетической диагностики (ПГД), она используется в сочетании с экстракорпоральным оплодотворением, её основная задача – помощь родителям-носителям в рождении здоровых детей.
Разновидности альфа-талассемии
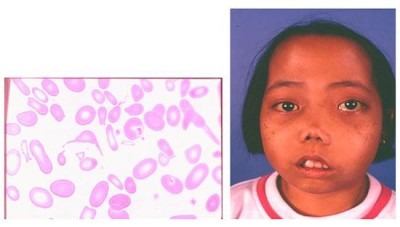 Silentcarrierstateили альфа-талассемия минор (малая альфа-талассемия). Состояние не влияет на здоровье. Недостаток альфа-белка невелик настолько, что не вызывает нарушений функций гемоглобина. Талассемия называется малой (silentcarrier– тихий носитель) из-за затруднительной постановки диагноза. Он ставится родителю, если у ребенка есть проявления гемоглобинопатии H.
Silentcarrierstateили альфа-талассемия минор (малая альфа-талассемия). Состояние не влияет на здоровье. Недостаток альфа-белка невелик настолько, что не вызывает нарушений функций гемоглобина. Талассемия называется малой (silentcarrier– тихий носитель) из-за затруднительной постановки диагноза. Он ставится родителю, если у ребенка есть проявления гемоглобинопатии H.
Гемоглобинопатия ConstantSpring (CS). Форма предыдущего состояния, вызванная изменением альфа-глобина. Название ConstantSpring получила в честь региона Ямайки, где впервые была выявлена. Пациент с CS не ощущает симптомов этого типа, осложнений нет.
Альфа-талассемия минор (minor, малая альфа-талассемия). Значительный недостаток альфа-глобина. Маленький размер эритроцитов и слабовыраженные признаки анемии – частые симптомы этого типа.
Гемоглобинопатия H. Значительный дефицит альфа-глобина, вызывающий тяжелую анемию и серьезные осложнения (спленомегалия селезенки, деформация конечностей). Её назвали в честь гемоглобина H, разрушающего эритроциты.
Гемоглобинопатия Н Constant Spring. Тяжёлая форма. У пациентов отмечены серьёзные признаки анемии. Этому состоянию присущи очень длинные цепи гемоглобина.
Гомозиготная талассемия Constant Spring. 2 постоянных носителя (родители) гемоглобинопатии HCS передают гены талассемии ребенку (в отличие от гемоглобинопатии H CS, при которой один родитель – носитель гемоглобинопатии HCS, а другой – переносчик альфа-талассемии минор).
Водянка плода (альфа-талассемия майор, major). Альфа-белки полностью отсутствуют. Гамма-глобины, производимые плодом, образуют аномальный гемоглобин Barts. Большая часть детей с этим заболеванием умирают еще до родов. В крайне редких случаях переливания крови позволяли родить ребёнка талассемией майор, когда заболевание было диагностировано в утробе, но новорожденный с таким диагнозом в дальнейшем требует постоянных переливаний.
Разновидности бета-талассемии
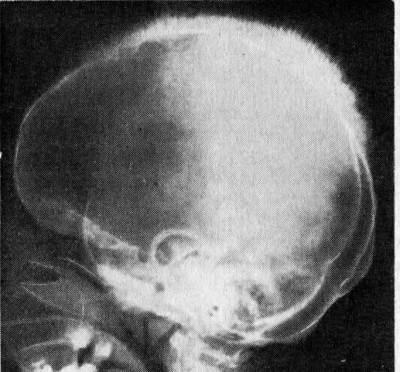 Талассемии минор (minor, малая талассемия). Малое количество бета-белка не провоцирует никаких осложнений. Человек является носителем, симптомов, как правило, нет, кроме легкой анемии. Специалисты часто расценивают маленький размер эритроцитов как признак железодефицитной анемии, что в корне неверно.
Талассемии минор (minor, малая талассемия). Малое количество бета-белка не провоцирует никаких осложнений. Человек является носителем, симптомов, как правило, нет, кроме легкой анемии. Специалисты часто расценивают маленький размер эритроцитов как признак железодефицитной анемии, что в корне неверно.
Талассемии интермедиа (intermedia). Дефицит бета-белка велик, это способно вызвать симптомы тяжелой анемии, а также серьезные осложнения (спленомегалию селезенки). Существует широкий диапазон клинической тяжести этого состояния, между талассемией интермедиа и талассемией майор довольно размытые границы, что может привести к ошибкам в диагнозе. Решающее значение играет число переливаний крови. Насколько сильно пациент зависит от переливаний, настолько больше будет вероятность, что его состояние граничит с талассемией майор.
Талассемия майор (major, анемия Кули). Самая тяжелая форма. Абсолютное отсутствие бета-белка способствует развитию анемии, несущей угрозу для жизни. Анемия Кули требует постоянных переливаний. Эти регулярные переливания крови (чаще на протяжении всей жизни) приводят к тому, что в организме скапливается избыток железа. Задача врачей – держать эти “перегрузки” под контролем с помощью терапии по удалению избытков железа во избежание отказа органов.
В дополнение к этим двум типам существуют другие родственные заболевания.E Бета талассемия. Гемоглобин Е – распространенный аномальный гемоглобин. На фоне Е бета талассемии развивается тяжелая анемия.
Серповидная-бета-талассемия. Комбинация гемоглобина S с бета талассемией. Гемоглобин Sвстречается у людей с серповидно-клеточной анемией (эритроциты изменённой формы — дрепаноциты). Этот вид анемии наследуется как доминантный признак.
Различия типов талассемии в виде таблицы:
α-талассемия:
| Недостающие альфа-гены | Расстройство | Признаки | Другие названия |
| 1 | носитель (silent carrier) | отсутствуют | aльфа-талассемия минима, alpha thalassemia minima |
| 2 | минор | лёгкие | aльфа-талассемия минор, alpha thalassemia minor |
| 3 | гемоглобинопатия H | средние | промежуточная альфа-талассемия, Hemoglobin H disease |
| 4 | майор | тяжёлые | водянка плод, альфа-талассемия майор, alphathalassemiamajor |
β-талассемия:
| Пораженные бета-гены | Расстройство | Признаки | Другие названия |
| 1 | носитель (silent carrier) | лёгкие | b талассемия минор, Beta thalassemia minor |
| 1 | минор | лёгкие | |
| 2 | интермедиа | средние | |
| 2 | майор | тяжёлые | анемия Кули, Cooley’s anemia |
Прогноз течения болезни
Пациенты с умеренной и тяжелой формой имеют хорошие шансы на выживание, их главная задача – придерживаться правильной программы лечения (трансфузионная терапия и профилактика уровней железа с помощью комплексообразующих агентов). Сердечно-сосудистые заболевания из-за избытка железа — основная причина смерти. Трансплантация костного мозга способна излечить больного. Пациентам с талассемией, возможно, потребуется хирургическое вмешательство, чтобы исправить скелетные деформации.
Репортаж про ученных, которые научились бороться с данным заболеванием
Больным требуется постоянная диагностика общего анализа крови и уровней железа. Важно проверять печень (из-за развития гемохроматоза) и сердце, сдавать анализы на вирусные инфекции.При беременности
Женщины с талассемией имеют высокий уровень бесплодия. Тем не менее, некоторые из них могут забеременеть.
- Если у одного из партнеров есть ген талассемии, есть большая вероятность, что ребенок унаследует болезнь;
- Стресс во время беременности только ухудшит симптомы болезни. Сердце и печень женщины являются наиболее уязвимыми во время беременности, очень важная задача – контроль эндокринной системы;
- Во время беременности объем крови в организме значительно возрастает. Это может привести к анемии, которая увеличит потребность в переливании крови. Чем выше объем крови в организме матери, тем больше нагрузка на её сердце;
- Женщины с талассемией имеют повышенный риск развития сахарного диабета 1 типа;
- Во время беременности женщине потреблять достаточное количество фолиевой кислоты, она поможет снизить риск развития мегалобластной анемии.
Заболевание у детей
 У некоторых детей с легкой формой талассемии симптомы могут отсутствовать. Но обычно больные дети чувствуют себя усталыми, раздраженными, у них частая одышка, головокружения, бледная кожа, бледные (синие) губы.
У некоторых детей с легкой формой талассемии симптомы могут отсутствовать. Но обычно больные дети чувствуют себя усталыми, раздраженными, у них частая одышка, головокружения, бледная кожа, бледные (синие) губы.
В более тяжелых случаях, у детей наблюдаются:
- Частый пульс;
- Увеличенная печень или селезенка;
- Желтуха;
- Деформация лобной кости;
- Замедленный рост (из-за анемии).
Дети также могут быть диагностированы до рождения, путем проверки образца амниотической жидкости или клеток из плаценты. Дети сдают анализы в том же порядке, что и взрослые, в том числе, анализ крови, анализ на гемоглобин.
Профилактика
- Переливание крови – введение эритроцитов, чтобы восстановить их уровень. Переливания проводятся с периодичностью в каждые 4 месяца (для больных тяжелой талассемией), и каждые 3-6 недель у больных бета-талассемией. Иногда переливания могут потребоваться больным гемоглобинопатией Hили бета-талассемией интермедиа;
- Удаление избытка железа из организма. Опасность множественных переливаний – рост уровня железа в организме, что может стать причиной болезней сердца. Лекарственное средство, например, жидкий дефероксамин вводят под кожу, или больной принимает таблетки, например, Deferasirox. Таким пациентам необходимо принимать добавки с фолиевой кислотой для восстановления эритроцитов;
- Спленэктомия (удаление селезенки) — вынужденная мера для пациентов с гемоглобинопатией Н, если их потребность в переливании постоянно растет;
- Трансплантация костного мозга (и стволовых клеток) от совместимого донора – единственный метод, способный излечить больного. Пересадка костного мозга от родственника даст лучшие результаты. Уже в течение двух недель пересаженные стволовые клетки начнут производить новые, здоровые клетки крови.
Образ жизни при данном заболевании
Важно:
- Стоит задача избегать потребления большого количества железа. Не стоит принимать витамины или другие добавки, содержащие железо, не проконсультировавшись с лечащим врачом;
- Ешьте здоровую пищу. Сбалансированный ежедневный рацион поможет почувствовать себя лучше. Лечащий врач может рекомендовать добавки с фолиевой кислотой (способствует созданию новых эритроцитов). Чтобы сохранить здоровье костей, рацион должен содержать оптимальное количество кальция и витамина D;
- Избегайте инфекций (в особенности после спленэктомии). Защитить себя от инфекций можно с помощью частого мытья рук и избегая больших скоплений людей;
- Больным талассемией показано прививаться от гриппа, пневмококка и гепатита B, важна диагностика показателей крови.
Группы риска
Частота возникновения альфа талассемии среди белых людей низкая. Около 15% афроамериканцев являются её носителями, и только 3% имеют слабо выраженные признаки. Гемоглобинопатия H редко встречается в этой группе населения. В Северной Америке из-за присутствия большого количества мультинациональных слоёв общества частота заболевания синдромом альфа талассемии увеличилась. У некоторых этнических групп (население Юго-Восточной Азии и средиземноморского населения) гемоглобинопатия H встречается чаще. Высокая частота проявления гемоглобинопатии Constant Spring (CS) была отмечена в Юго-Восточной Азии.
Наследственность
Это заболевание наследуется как не полностью доминантный аутосомный признак от родителей к детям через мутировавшие гены гемоглобина.
Если в семье присутствовали случаи талассемии, ребенок имеет повышенный риск развития заболевания.Международная статистика
Альфа-талассемия – наиболее распространённое генетическое заболевание в мире. Существует 270 миллионов носителей мутировавших генов глобина, которые могут передать тяжелые формы талассемии по наследству. Кроме того, 300000-400000 больных младенцев рождаются каждый год, более 95% из которых находятся в Азии, Индии или на Ближнем Востоке.
Частота проявления этой болезни составляет 20-30% в частях Западной Африки, 60-80% — в некоторых районах Саудовской Аравии, Индии, Таиланда, Папуа-Новой Гвинеи и Меланезии. В Таиланде около 7000 детей ежегодно рождается с гемоглобинопатией H. Среди китайского населения диапазон больных составил от 5% до 15%. Частота возникновения альфа-талассемии в Великобритании, Исландии и Японии ниже 0,01%.
Пол
Мужчины и женщины затронуты в равной степени.
Интересные факты о болезни
- Альфа-талассемия встречается в странах, эндемичных по малярии. Исследования показали, что наличие одиночных и двойных генов альфа-глобина оказывает защитное действие против малярии;
- Люди рождаются с этим расстройством. Единственным исключением из этого правила являются пациенты с миелодиспластическим синдромом – это пожилые люди, средний возраст на момент постановки диагноза – 68 лет;
- По данным Центров по контролю и профилактике заболеваний талассемия является наиболее распространенным заболеванием среди людей из Азии, Ближнего Востока, Африки и средиземноморских странах (85%), таких как Греция (4%) и Турция (7%). Отсюда название болезни средиземноморская анемия;
- Существует тип альфа-талассемии, связанный с умственной отсталостью, который затрагивает только мужчин. Это состояние называется синдром альфа-талассемии и умственной отсталости.